mut 分析
前言
IMPORTANT
mut 模块专注于单细胞 SNV/Indel 富集分析,通过突变矩阵与表达矩阵的联合建模,定位携带特定突变的细胞群并评估其功能通路。流程默认承接上游完成的变异检测结果(*.snp_indel.all_UMI.matrix、*.snp_indel.alt_UMI.matrix),无需在本模块内重复调用 VarScan 等长流程。
随着单细胞测序广泛应用,很多研究希望回答“某个细胞群是否携带特定突变并呈现功能增益”。SeekSoul Online 云平台的 mut 模块正是为此打造:它能自动完成矩阵校验、样本拆分、突变富集、差异/通路分析与报告生成,大幅降低生信开发门槛。
mut 分析理论基础
核心原理
- 矩阵校验与下载:系统会自动读取
sample_matrix.txt中记录的all_UMI/alt_UMI路径,完成可用性校验并并行下载,确保每个样本的突变矩阵就绪。 - RDS 子集与样本识别:根据用户指定的样本列(默认
Sample)与细胞注释列生成子集 RDS,为后续分析统一 barcode 命名并过滤无关细胞。 - 突变信息汇总:流程读取突变矩阵,统计每个位点的 UMI 数、携带突变的 barcode 数、突变率以及出现在不同 celltype/cluster 中的情况。
- 突变富集判定:将突变矩阵挂载为单细胞对象的额外 Assay,对每个位点 × 细胞群使用 Fisher 精确检验 判断是否显著富集,并输出
*_snv_markers.xls与 UMAP 可视化。 - 差异与通路分析:若物种为人/小鼠,系统会自动挑选前 10 个显著位点,对其突变细胞 vs 覆盖细胞执行差异分析,并串联 GO/KEGG/Reactome 富集,输出表格与图像。
- 报告生成:生成可直接交付的 HTML/PDF 报告。
单样本 vs 多样本策略
| 场景 | 处理思路 | 产出 |
|---|---|---|
| 单样本 | 直接针对该样本的 *.snp_indel.*.matrix 进行统计、富集与可视化 | Sample.mut.info.txt、mutation_umap/、差异富集(若 species ∈ {human, mouse}) |
| 多样本 | 系统会生成 multi(所有突变)与 common(公共位点)两套矩阵,并分别执行统计与富集,便于比较整体与交集 | multi.* 与 common.* 双份结果,并在报告中分章节展示 |
关键统计指标
- UMI / barcode:反映突变在细胞层级的覆盖度,可用于评估测序深度是否足够。
- mut_rate:
barcode_count / total_cells,衡量突变在该样本中的频率。 - Fisher 精确检验:将“突变 vs 覆盖”与“目标细胞 vs 其他细胞”构成 2×2 列联表,返回
p_val、ident1_mut等指标。 - 差异表达/富集:默认
logfc.threshold=0.25,GO/KEGG/Reactome 统一绘图并输出表格。
SeekSoul Online 云平台操作指南
分析前准备
CAUTION
- 上游突变矩阵文件应与 RDS 内的 barcode 命名保持一致;如包含后缀,mut 流程会自动匹配,但仍建议在上传前自检。
- metadata 的列名与内容请勿包含中文或特殊字符(
&、空格等),否则流程可能失败。
- 仅当
species设为 human/mouse 时才会执行差异富集模块。
参数详解
| 界面参数 | 说明 | 备注 |
|---|---|---|
| 任务名称 | 英文开头,可含中文/数字/下划线 | 用于报告抬头与任务跟踪 |
| 分组因子 | metadata 中代表样本的列,默认 Sample | 决定 subset_samples.R --group |
| 细胞类型 | metadata 中的细胞注释列,如 CellAnnotation | 影响富集检验与差异分析 |
| 样品类型 | 要分析的样品信息,及对应 all_UMI.matrix 和 alt_UMI.matrix。 | 支持 OSS 路径 |
| 物种 | human / mouse / other | 控制是否执行差异富集 |
| 备注 | 自定义文本 | 记录分析背景 |
结果解读
结果目录速览
| 路径 | 内容 | 说明 |
|---|---|---|
output/results/<sample>.mut.info.txt | 每个位点的 UMI、barcode、mut_rate 及 cluster 信息 | 可用于下游筛选热点突变 |
output/results/<sample>/mutation_umap/SNV_diff/*.png | Fisher 显著突变的 UMAP 可视化 | 图名即突变位点 |
output/results/<sample>/mutation_umap/SNV_diff/<sample>_snv_markers.xls | 突变富集统计表 | 含 p_val、ident*_mut/cover 等 |
.../diff_pathway/pos*/diffgene.xls | 差异表达结果 | ident.1 = alt,ident.2 = WT |
| `.../diff_pathway/pos*/go | kegg | reactome/` |
report/ | HTML 报告目录 | 打包 report.zip 供下载 |
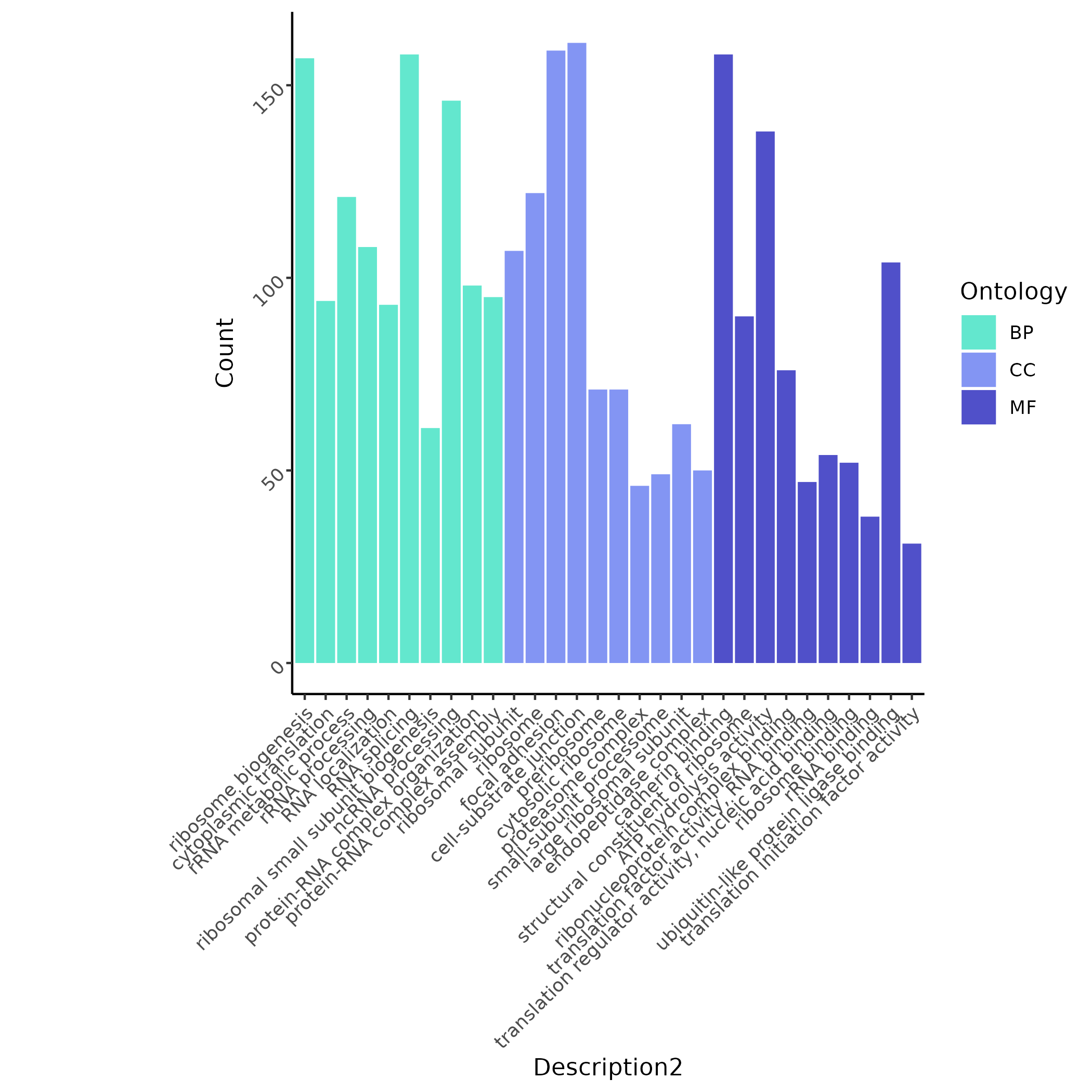
关键图表示例
单样本视图
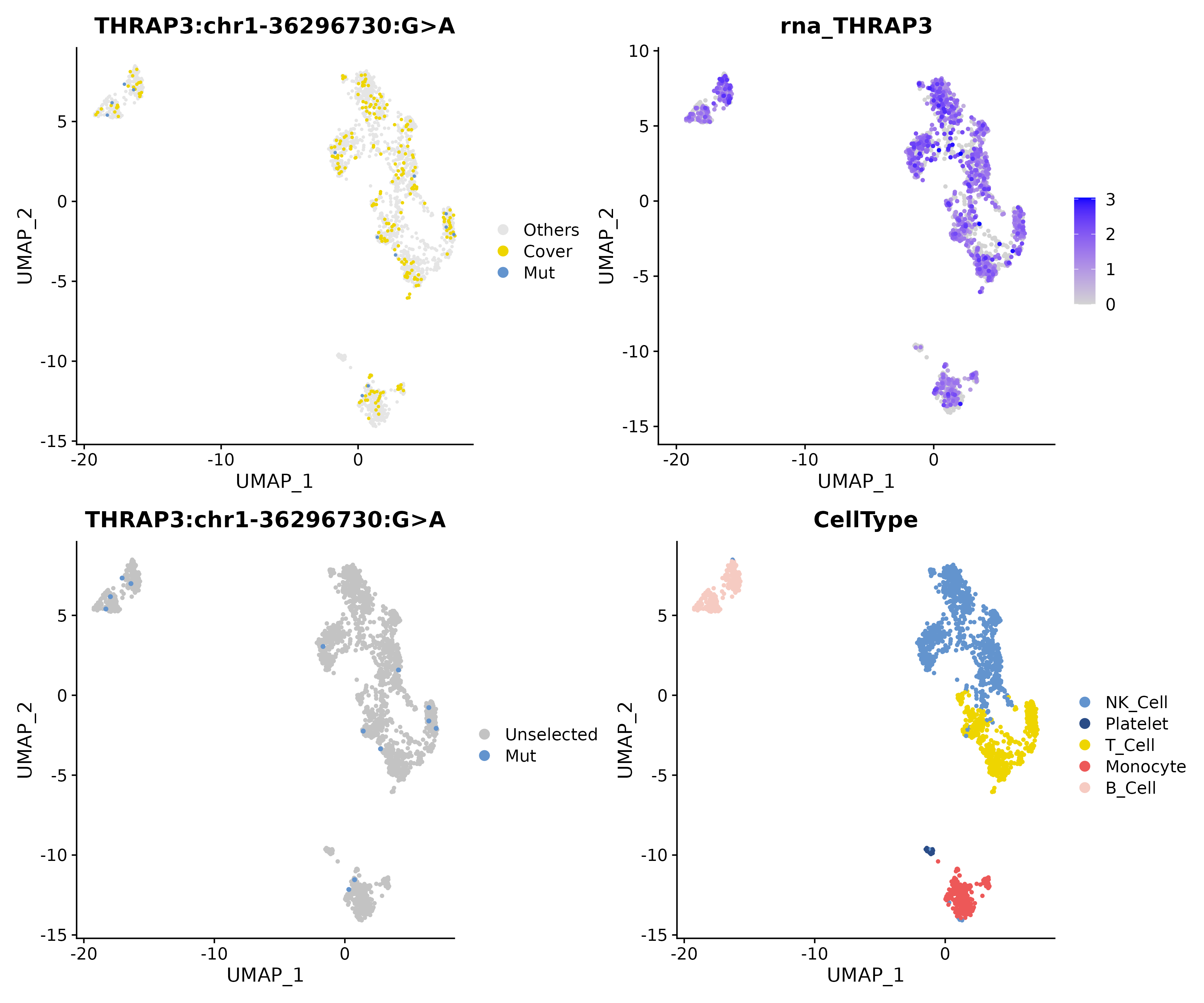
展示 Fisher 检验筛选出的显著位点(示例:PBMC 样本中 B 细胞上调的
THRAP3 chr1-36296730 G>A)。红色为突变细胞,灰色为覆盖但未突变的细胞。
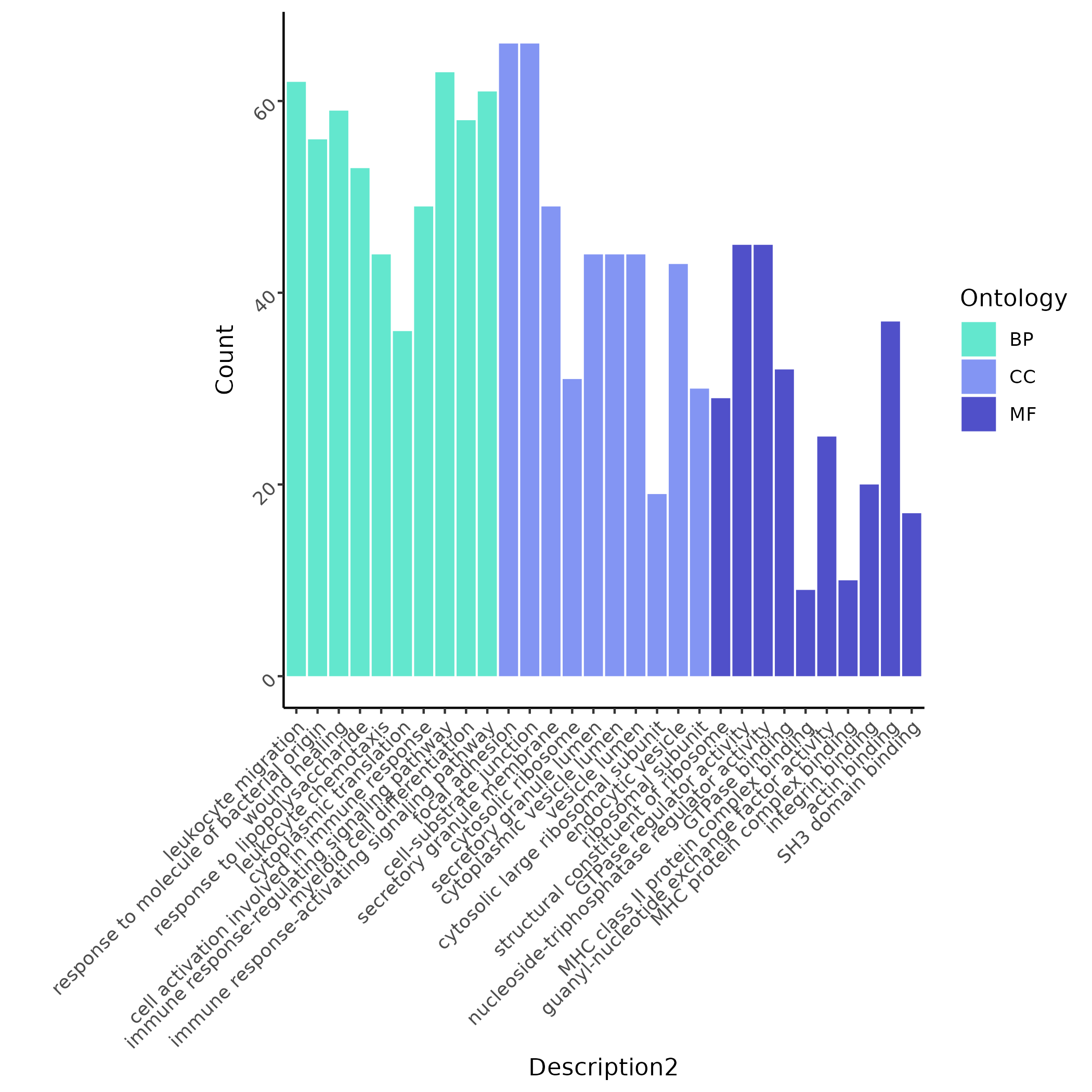
系统在
pos0EGR1位点的突变 Monocyte vs WT 细胞差异分析基础上,筛选显著项绘制柱状图,可快速定位“leukocyte migration”“wound healing”等主题。
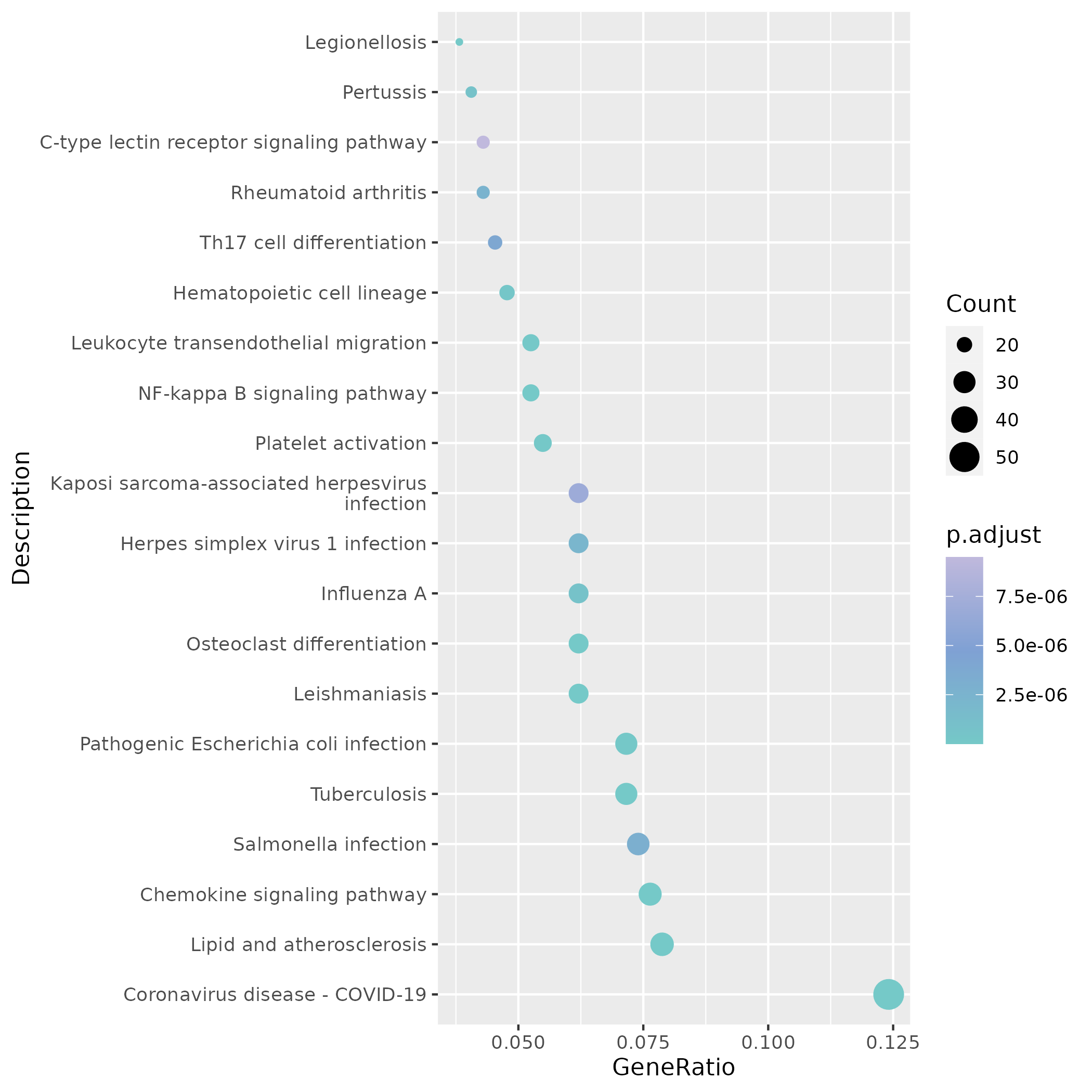
KEGG 富集散点图聚焦于免疫/感染相关通路(Chemokine signaling、NF-κB、Platelet activation 等),点的颜色与大小分别代表显著性与富集基因数。
多样本视图
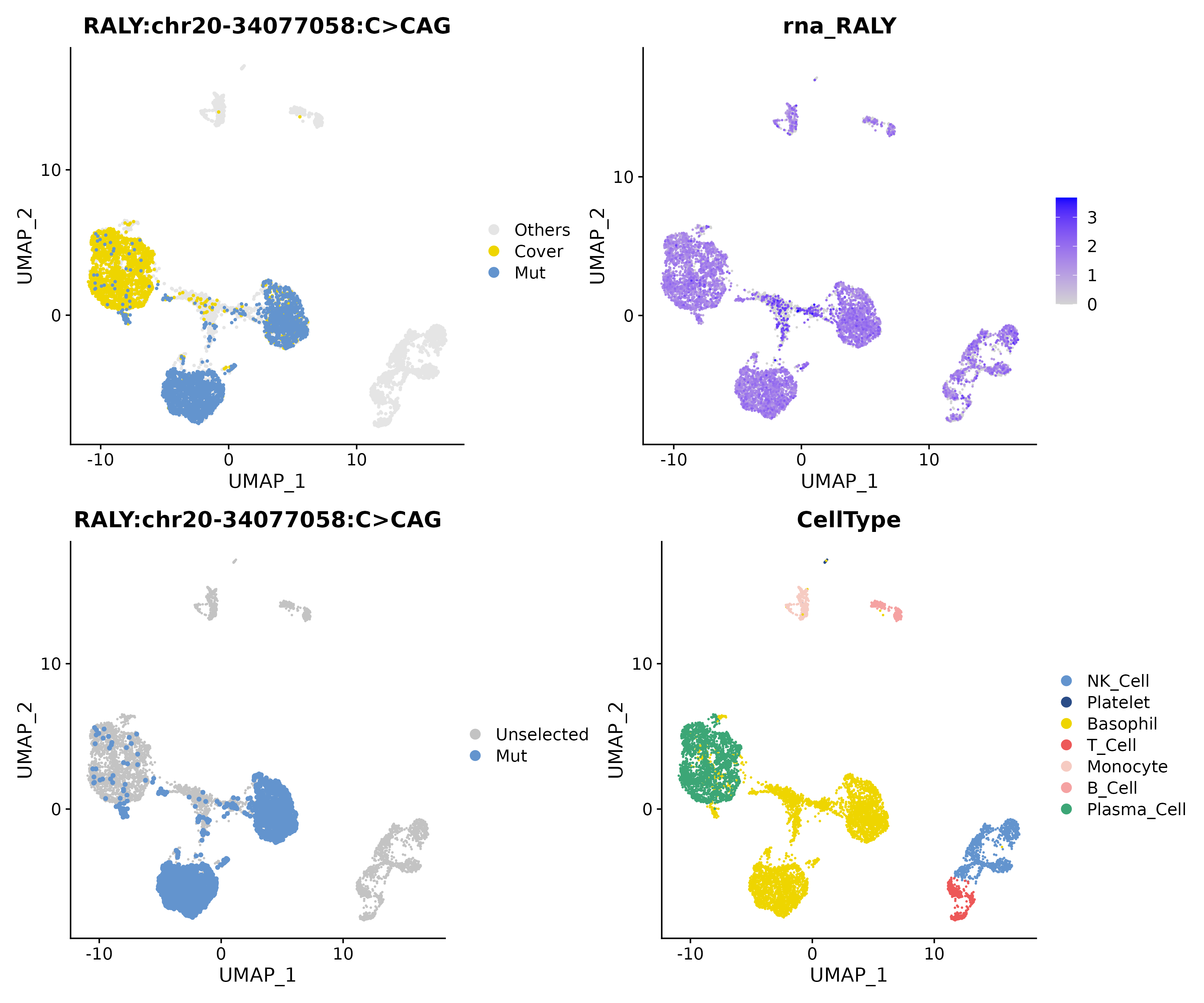
multi矩阵下,Basophil 细胞中RALY chr20-34077058 C>CAG显著富集,图中红色为突变细胞,灰色为覆盖细胞。
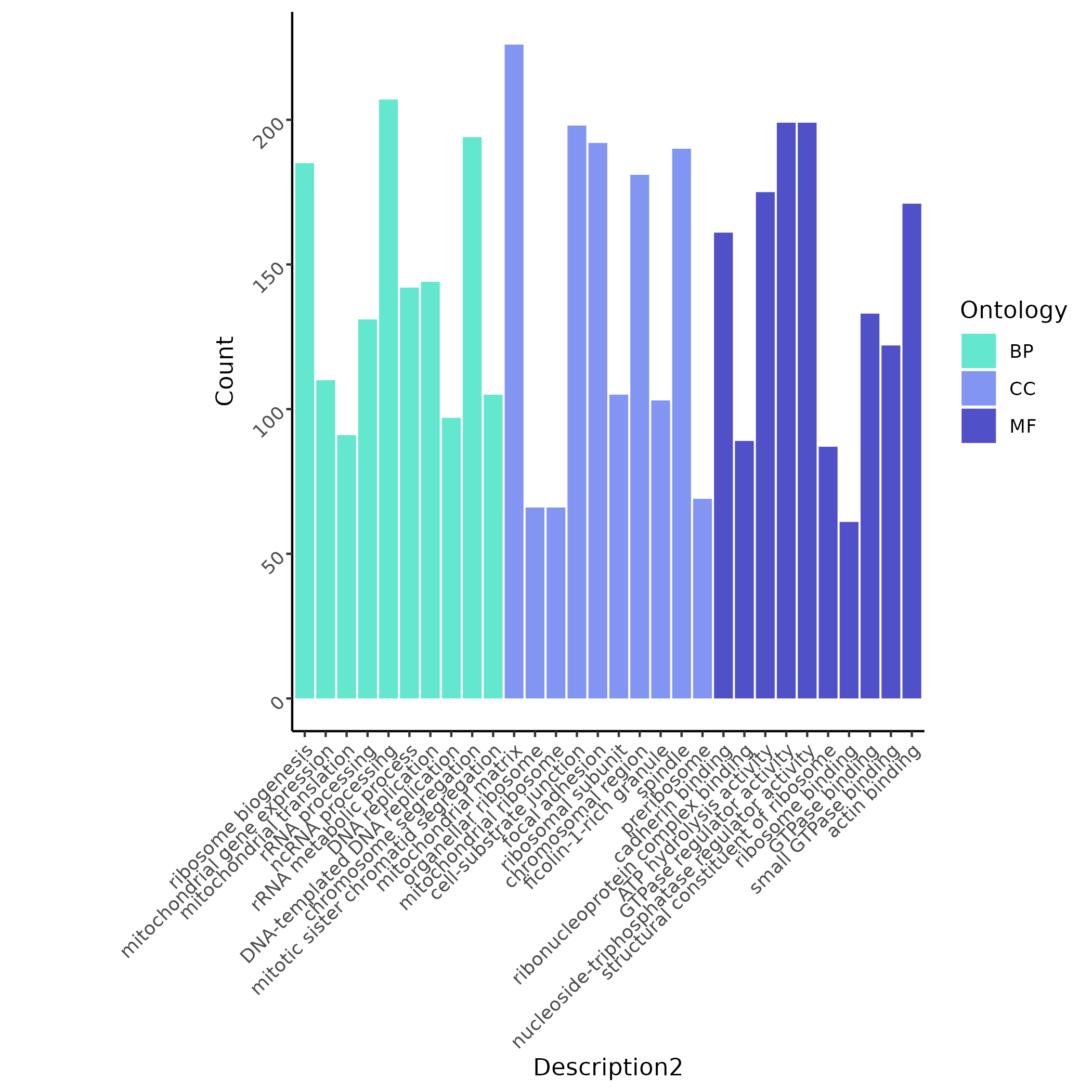
多样本差异分析显示 RALY 突变 Basophil 富集 ribosome biogenesis、mitochondrial gene expression 等核糖体/线粒体过程。
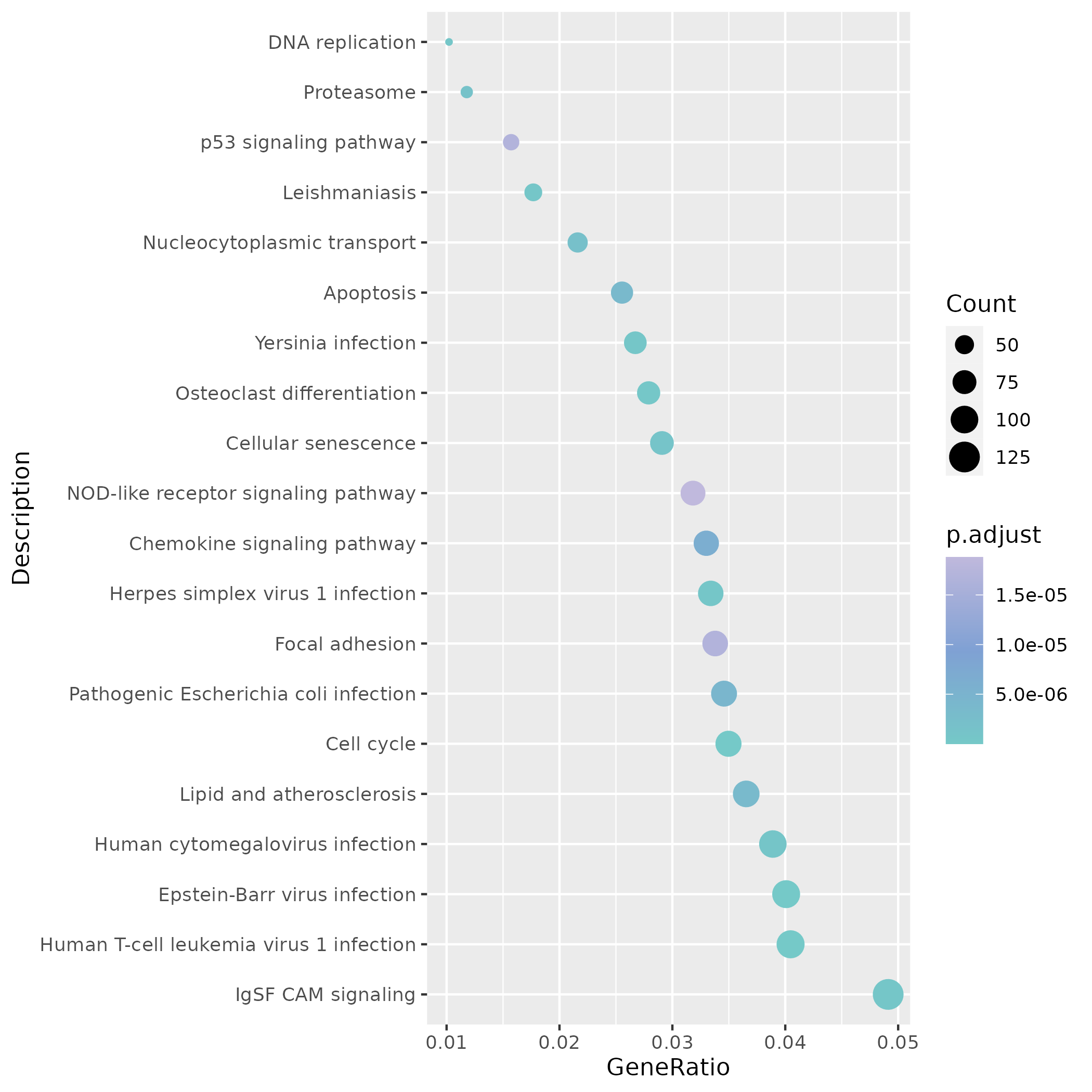
KEGG 方面,同一位点突出 DNA replication、Cell cycle 等增殖相关通路,提示该突变细胞具有高合成活性。
共同变异视图
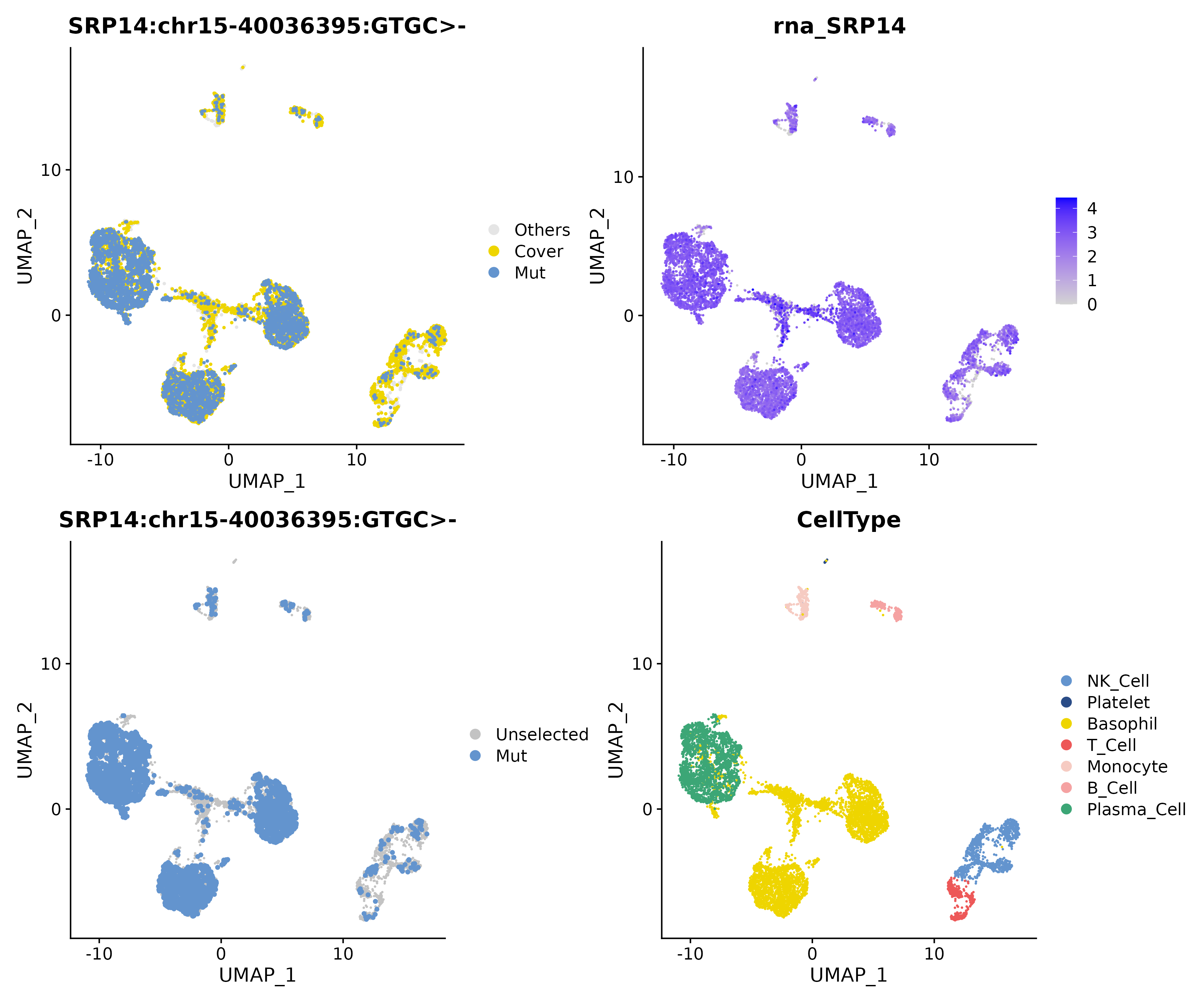
common矩阵强调所有样本共同存在的SRP14 chr15-40036395 GTGC>-,在 Plasma Cell 中呈现一致富集。
SRP14 突变相关细胞主要富集在 ribosome biogenesis、RNA processing 等转录/翻译流程。
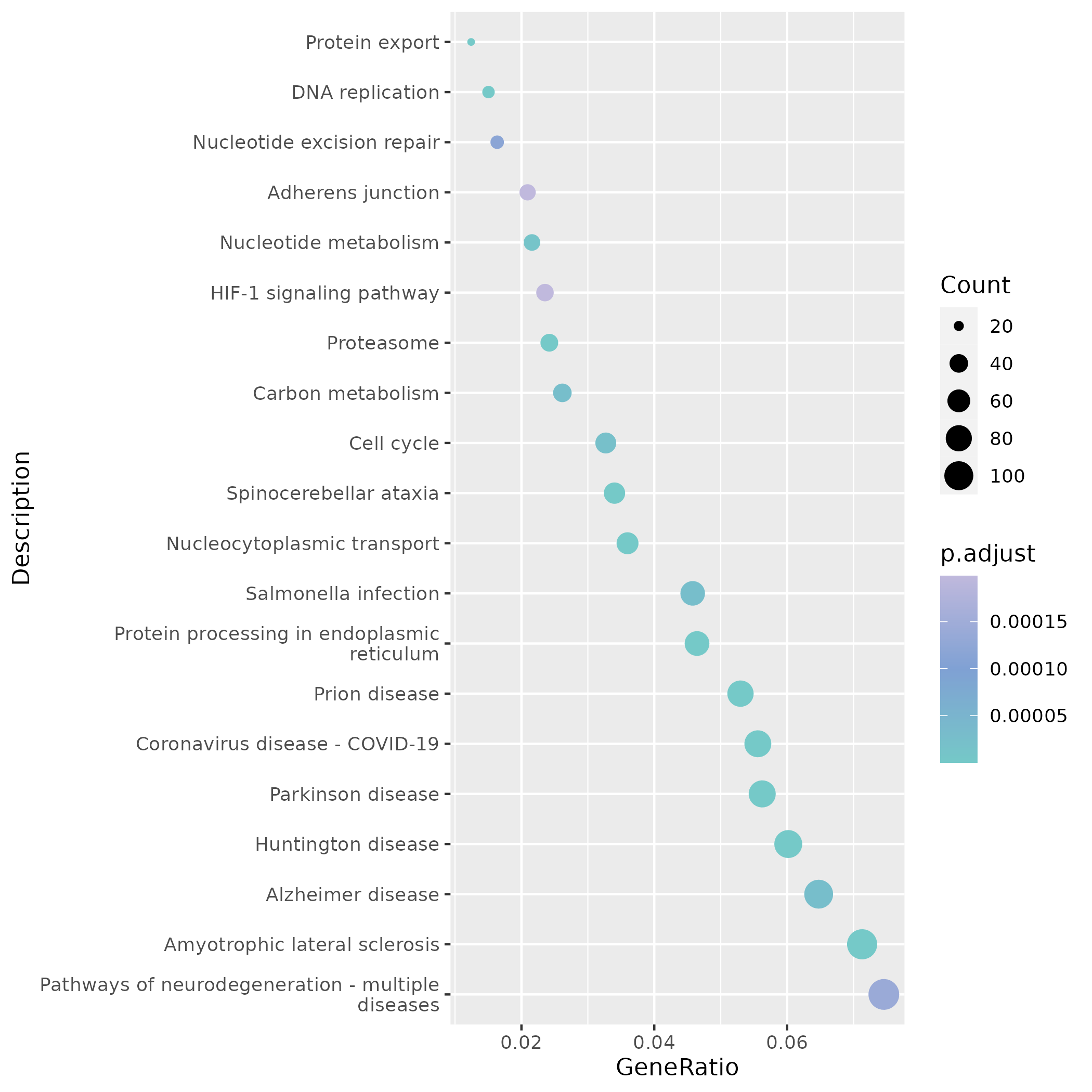
KEGG 结果强调 Ribosome、Spliceosome 等基础分子机器,加深了对共同突变功能背景的理解。
案例参考:最新单细胞突变实践
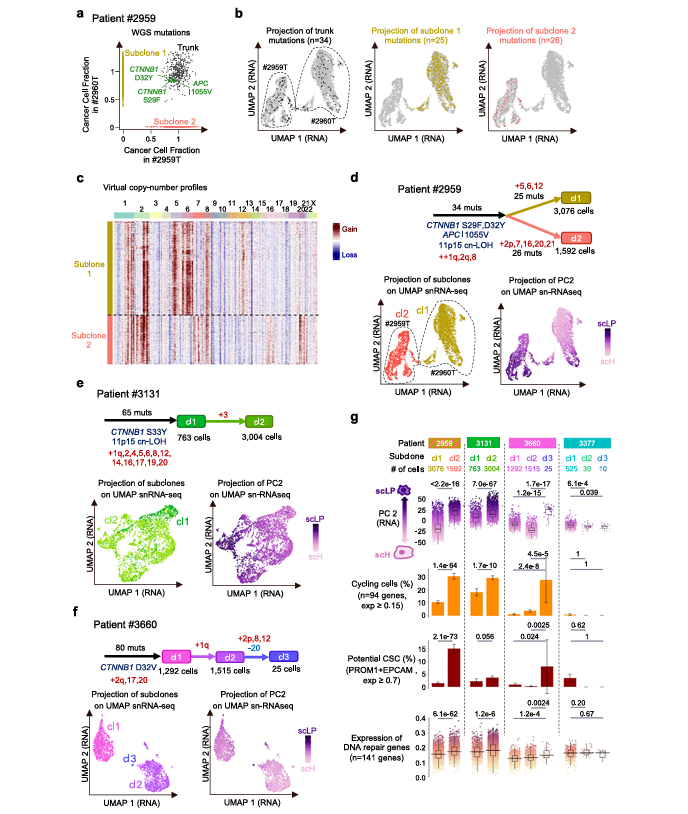
mut 模块的流程与近年的高影响力研究一致。以 Roehrig A 等人发表在 Nature Communications(2024, 15:3031)的肝细胞母细胞瘤(HB)单细胞多组学研究为例:
- 肝细胞母细胞瘤克隆演化与化疗响应研究
- Roehrig A 等在研究中,通过单细胞多组学(snRNA-seq + snATAC-seq)结合全基因组测序(WGS),实现了 HB 肿瘤单细胞水平的克隆架构重建与突变定位 —— 这与 mut 模块 “突变 - 细胞群 - 功能” 的分析逻辑高度契合。研究首先利用 WGS 识别 HB 关键驱动突变(如 CTNNB1 激活突变、11p15.5 位点拷贝中性杂合性缺失 cnLOH),再通过单细胞数据将这些突变映射到具体细胞亚群,明确每个遗传亚克隆的分化状态范围(如 scH 肝细胞样、scLP 肝祖细胞样、scM 间充质样)。
- 类似地,在 mut 模块分析中,可通过 Fisher 精确检验定位显著富集特定突变的细胞亚群(对应文献中 “亚克隆分化状态分析”)。例如,若在 HB 样本中检测到 scLP 亚群显著富集 CTNNB1 突变,可进一步对该突变细胞群与野生型细胞群进行差异表达分析,通常能观察到文献中提及的 “干细胞标志物(如 PROM1)与 DNA 修复基因高表达” 特征;后续结合 KEGG 通路富集,还可验证这些突变是否激活细胞周期、DNA 修复相关通路(如文献中 scLP 亚克隆化疗后增殖更快的功能关联),从而揭示突变对肿瘤细胞化疗耐药性的影响机制。
推荐的实践路径是:
- 突变定位:利用
*_mut.info与SNV_diff识别显著富集在特定 celltype/cluster 内的 SNV。 - 功能评估:对这些位点进行差异分析 + GO/KEGG/Reactome 富集,观察它们是否集中于细胞周期、免疫通路或代谢通路。
- 结果交付:借助报告模块导出图片与表格,将"突变–细胞类型–功能通路"的链条串联起来,写入项目报告或论文。
通过这种方式,我们可以在单细胞分辨率上理解肿瘤的异质性,为精准医疗提供更深入的见解。
注意事项与最佳实践
WARNING
mut 流程不执行变异检测,只分析上游产出的矩阵;若矩阵质量差或样本 barcode 不匹配,将直接影响富集结果。
- 合理筛选样本:单细胞样本差异较大,建议优先选择有足够覆盖(≥3k 细胞、UMI 深度>20k)且 meta 注释准确的项目。
- 多样本解读:multi 与 common 结果含义不同——前者展示所有突变,后者强调“跨样本一致”的热点;报告已分章节呈现。
常见问题(FAQ)
Q:为何提示"突变矩阵与 RDS barcode 不匹配"?
A:通常是上游矩阵保留了_1、-1等后缀。mut 流程会尝试匹配,但若完全不重合(setdiff=all)则会报错。请确认矩阵列名是否与 Seurat 对象一致或可通过后缀匹配。Q:差异富集没有结果?
A:需要满足两个条件:species ∈ {human, mouse}且SNV_diff至少存在一个p_val < 0.05的位点。可在上传参数时确认物种或放宽group_input_name,以获得更多显著位点。Q:报告中"共同变异"章节为空?
A:多样本项目若不同样本之间没有公共位点(common_alt_pos为空),则com_mut章节只会显示提示信息。可检查是否所有矩阵都共享同一pos列。Q:如何自定义富集数据库?
A:默认使用org.*.eg.db+ Reactome + KEGG(本地镜像)。若需替换,可在工作空间中更新mut.go_enrich1.R/mut.kegg_enrich1.R的参数,但当前云平台界面尚未开放该配置。
参考资料
[1] SKINNIDER M A, SQUAIR J W, KATHE C, et al. Cell type prioritization in single-cell data[J]. Nat Biotechnol, 2021, 39(1): 30-34.
[2] KATHE C, SKINNIDER M A, HUTSON T H, et al. The neurons that restore walking after paralysis[J]. Nature, 2022, 611(7936): 540-547.
[3] ROEHRIG A, et al. Single-cell multiomics reveals the interplay of clonal evolution and cellular plasticity in hepatoblastoma[J]. Nature Communications, 2024, 15: 3031.